2021年7月23日,张宏研究员联合法国巴黎大学的Patrice Codogno教授以及南方科技大学生命科学院的赵燕教授在国际著名期刊、影响因子高达94.444分的Nature Reviews Molecular Cell Biology杂志上发表题为《Machinery, regulation and pathophysiological implications of autophagosome maturation 》的综述文章。此文章全面概括了近年来在自噬小体成熟分子机制,调控以及病理生理学意义方面的研究进展。

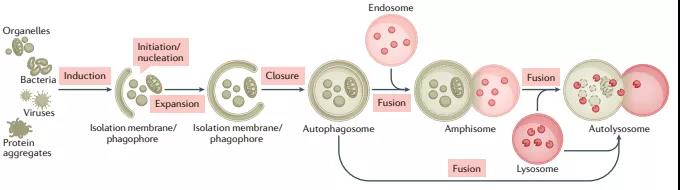
自噬是一种用于维持细胞稳态的多功能降解系统,其中细胞质材料被隔离在双膜自噬小体中,随后被输送到溶酶体,在那里它们被分解。在多细胞生物中,新形成的自噬小体经历一个称为“成熟”的过程,在这个过程中,它们与源自内溶酶体区室(包括早期/晚期内吞体和溶酶体)的囊泡融合,形成两性体,最终成为降解性自体溶酶体。这种融合过程需要多个膜动力学调节剂的协同作用,包括SNARE、束缚蛋白和RAB GTP酶,以及自噬小体和晚期内吞体/溶酶体相互运输。
多种机制调节自噬小体的成熟,包括关键成分的翻译后修饰、膜上磷酸肌醇脂质种类的空间分布、RAB蛋白动力学以及溶酶体的生物发生和功能。营养状态和各种应激整合到自噬小体成熟的机制中,以协调自噬通量的发展。自噬小体成熟障碍与多种人类疾病的发病机制有关,包括神经退行性疾病、癌症和肌肉疾病。此外,入侵的病原体利用各种策略来阻止自噬小体的成熟,从而逃避破坏甚至颠覆自噬泡(自噬小体、两性体和自噬溶酶体)以使其存活、生长或释放。
在这里,我们讨论了对自噬小体成熟机制和调节的最新进展,这些机制与人类病理生理学的相关性以及病原体如何利用它们来获益。我们还提供了治疗靶向自噬小体成熟的观点。
多细胞生物体中的细胞不断承受各种应激,包括蛋白质错误折叠、细胞器损伤、营养能量缺乏和病原体入侵。细胞用来对抗应激的一种机制是溶酶体介导的细胞内成分通过自噬降解。自噬涉及形成隔离膜(或吞噬细胞),该膜进一步膨胀和闭合形成双膜自噬小体。
自噬小体形成机制近期研究进展:各种细胞材料可以隔离在自噬小体中,包括未选择的细胞质材料或选择的传递物质,如蛋白质聚集体、受损的细胞器和病原体。在通过膜脱落关闭自噬小体后,“货物”被输送到液泡(酵母和植物)或溶酶体(动物细胞)。自噬物降解后,自噬溶酶体中消化的内容物被释放,溶酶体重新形成以维持自噬通量。由ATG(自噬相关)基因和EPG(异位PGL颗粒)基因编码的一系列蛋白质——主要分别从酿酒酵母和秀丽隐杆线虫的遗传筛选中鉴定——在不同的膜重塑步骤中作用于自噬小体的形成和成熟。
在酵母中,自噬小体在更大的液泡附近形成并与其直接融合,而在多细胞生物中,新形成的自噬小体与不同的内溶酶体囊泡(例如早期/晚期内吞体和溶酶体)融合,形成非降解的单膜结构称为“两性体”, 通过水解酶的积累逐渐获得降解特性。成熟还涉及由V-ATPase介导的自噬小体酸化,这是降解酶活性所必需的。
自噬小体与内吞体/溶酶体的融合需要同源SNARE(可溶性N-乙基马来酰亚胺敏感因子附着蛋白受体)复合物、栓系蛋白、磷酸肌醇和RAB蛋白的协同作用。自噬液泡(包括自噬小体、两性体和自体溶酶体)和晚期内吞体/溶酶体在细胞核周空间和细胞外围之间的双向微管运输使相遇频率和融合效率最大化。
最近的研究表明,调节自噬小体成熟的机制,如SNARE复合体组装、系链募集和RAB蛋白动力学,受到严格控制,并对营养可用性和应激条件高度敏感。扰乱这些过程会导致受损细胞器和有毒蛋白质聚集体的积累,也可能阻碍内吞运输。因此,受损的自噬小体成熟与各种人类疾病的发病机制有关,包括神经退行性疾病、癌症和肌肉疾病。此外,入侵的病毒和细菌还广泛靶向自噬小体的成熟以阻止其降解能力。病毒和细菌因此逃避破坏,并积累自噬液泡以供其生存、复制和释放。
值得注意的是,与自噬小体形成所必需的基因受损不同,作用于自噬小体成熟的基因功能丧失通常会导致较弱的缺陷和自噬液泡的逐渐积累。更重要的是,在某些情况下,可以通过促进部分冗余因素或机制的活动来抑制此类缺陷。例如,促进一种束缚因子的招募将减轻由另一种束缚因子耗尽引起的缺陷。这提供了改善相关人类疾病中自噬小体成熟缺陷的机会。
在这篇综述中,我们概述了我们对驱动和调节自噬小体成熟的分子机制的理解的最新进展。我们进一步讨论了自噬小体成熟受损与神经变性、肌肉疾病和癌症的发病机制之间的因果关系。我们还考虑了病毒蛋白和细菌效应子如何为了自身利益而阻止自噬体成熟。最后,我们提供了有关自噬体成熟如何在治疗上靶向对抗疾病的见解。
自噬小体的成熟需要它们与功能性内溶酶体区室(例如早期内吞体、晚期内吞体/多泡体和溶酶体)融合。因此,这些内溶酶体区室的生物发生紊乱——例如由ESCRT(运输所需的内体分选复合物)复合体和 COPI 囊泡的亚基功能丧失引起的紊乱——导致非降解性自噬液泡的积累。
自噬小体与晚期内吞体/溶酶体的融合涉及RAB蛋白、栓系蛋白和 SNARE复合物的协同作用。
膜融合由Qa、Qb、Qc 和 R SNARE组成的反式SNARE复合物的组装驱动。
通过小RAB GTP酶、磷酸肌醇和SNARE蛋白的活性形式募集到靶膜或融合囊泡的束缚因子促进囊泡的初始捕获和反式SNARE复合物的后续形成。
自噬液泡装饰有脂化(磷脂酰乙醇胺偶联)泛素样自噬蛋白,与酵母Atg8相关。
这些蛋白质,在LC3亚家族或GABARAP 亚家族中,还有助于募集其他因子,如系链和RAB鸟嘌呤核苷酸交换因子 (GEF) 以使其成熟。
LC3是一种广泛使用的自噬标记物,它在生物发生的不同阶段标记自噬结构,并且与晚期内吞体/溶酶体的标记物一起,它在成熟过程中积累在两性体上。
值得注意的是,在使用 LC3标记闭合后的自噬结构的研究中(可以通过与其他自噬蛋白的标记物共同标记或使用 HaloTag–LC3 方法将其与隔离膜区分开来),无法区分双膜自噬小体来自单膜两性体和自溶酶体。
因此,闭合后的LC3标记结构应称为“自噬液泡”,除非该结构已被明确证明。
由自噬小体膜定位的STX17(Qa)、SNAP29(Qbc)和晚期内吞体/溶酶体定位的VAMP8(苍蝇中的Vamp7和蠕虫中的VAMP-7)或自噬小体YKT6、SNAP29和晚期内吞体/溶酶体形成的SNARE复合物。局部STX7,部分冗余地驱动自噬小体与晚期内吞体/溶酶体融合(图1a)。
STX17靶向形成自噬体伴随自噬小体闭合。小鸟苷三磷酸酶IRGM通过与自噬小体膜上的STX17和ATG8样蛋白相互作用促进STX17易位至自噬小体。SNAP29是通过与其他 SNARE互动来招募的。自噬小体相关自噬蛋白ATG14促进STX17–SNAP29复合体组装,该蛋白直接与STX17相互作用(图1a)。
不同的SNARE复合物可能在自噬体成熟的不同阶段起作用,或驱动自噬泡与不同的晚期内吞体/溶酶体融合。融合后SNARE分解并返回其供体区室以保持细胞内膜的特性并为新一轮的融合做好准备。从自溶酶体中驱动SNARE 回收的分子机制尚未确定。已经表明,自溶酶体中STX17的释放与由于溶酶体酶分解导致的自噬体内膜的塌陷同时发生。
多亚基系链复合物和大的单个系链蛋白都参与自噬小体和两性体的初始捕获,并促进与早期/晚期内吞体和溶酶体的融合效率和特异性,但不与其他囊泡如或自噬体成熟过程中的分泌囊泡(图 1b)。
HOPS(同型融合和蛋白质分选)复合物是一种突出的束缚复合物,可促进反式 SNARE复合物的组装,充当RAB7的GEF并充当SNARE伴侣以促进自噬小体与晚期内吞体和溶酶体的融合。HOPS复合物可靶向自噬小体和晚期内吞体/溶酶体以促进融合。将HOPS复合物靶向晚期内吞体/溶酶体是通过其与RAB7和磷酸肌醇(如3-磷酸磷脂酰肌醇(PtdIns3P)35)的结合介导的,而多种机制促进其从细胞溶质到自噬体的易位,包括与STX17的相互作用、活性RAB7、GABARAPs38 或大系链蛋白(例如 PLEKHM1)如下所述(图 1b)。
EPG5是一种进化上保守的大系链蛋白,用于自噬小体成熟。它是一个RAB7效应子,通过与RAB7相互作用从细胞质转移到晚期内吞体/溶酶体(图 1b)。
在自噬小体成熟过程中,EPG5通过与LC3结合来捕获自噬小体/两性体并促进 STX17-SNAP29-VAMP8复合物的组装。EPG5耗竭导致自噬小体、两性体和非降解性自溶酶体的积累。EPG5在自噬小体成熟中的功能似乎需要其晚期内吞体/溶酶体定位。β-螺旋桨蛋白WDR45和WDR45B(酵母 PtdIns3P 结合自噬蛋白Atg18的哺乳动物同源物;也分别称为 WIPI4 和 WIPI3)与EPG5的晚期内吞体/溶酶体靶向相互作用并介导。在 Wdr45-Wdr45b双基因敲除细胞中,EPG5错误定位于自噬小体,EPG5-STX17相互作用增强。然而,STX17-SNAP29-VAMP8的组装减少,因此自噬小体成熟受损。
PLEKHM1,另一种束缚蛋白和RAB7效应器,优先与GABARAP亚家族成员相互作用以促进自噬小体成熟(图 1b)。PLEKHM1直接招募HOPS复合体,确保融合特异性和效率。溶酶体定位的pleckstrin同源(PH)结构域包含TECPR1选择性结合LC3C并促进LC3C修饰的自噬小体/两性体与 4-磷酸磷脂酰肌醇(PtdIns4P)富集的溶酶体(图1b)。还表明TECPR1在与自噬小体 PtdIns3P 结合后,采用与ATG12-ATG5偶联物相关的构象,从而促进自噬小体与溶酶体的束缚。
在非极化细胞中,自噬小体在整个细胞质中产生,而溶酶体主要位于核周区域。自噬小体的成熟需要
逆行运输
自噬泡,并通过微管马达蛋白动力蛋白驱动的溶酶体,以及它们的
顺行传输
由驱动蛋白介导的马达,其允许两个隔室,以满足和保险丝。
溶酶体的顺行运输使它们能够与外周自噬体融合。顺行转运由晚期内吞体/溶酶体相关多亚基
BORC复合物
介导,其募集小GTPase
ARL8
以促进ARL8依赖性偶联到驱动蛋白马达(图
2
)。
ARL8 直接与驱动蛋白 3 相互作用,或与其效应子 SKIP 相互作用,后者又与驱动蛋白 1 结合。BORC 复合物的消耗降低了外周自噬体与溶酶体的融合。RAB7效应子 FYCO1 还充当驱动蛋白1的接头,通过其与LC3和PtdIns3P
的
结合介导晚期内吞体/溶酶体和自噬液泡的顺行转运(图
2
)。
将动力蛋白-动力蛋白运动复合物募集到晚期内吞体/溶酶体和自噬液泡以进行逆行运输是由RAB7及其效应物RILP和ORP1L介导的(图
2
)。RILP与动力蛋白-动力蛋白运动复合物的p150亚基相互作用,而 ORP1L促进动力蛋白复合物与晚期内体/溶酶体膜相关βIII血影蛋白的结合。此外,ORP1L可以感知胆固醇水平并将其与溶酶体和自噬液泡的运输结合起来(图
2)
。
在低胆固醇条件下,ORP1L与内质网 (ER) 蛋白 VAPA 相互作用,形成ER与溶酶体/自噬泡之间的接触;然后将动力蛋白马达从RAB7-RILP解离,引起这些细胞器的扩散。在 Niemann-Pick C 型疾病和其他
溶酶体贮积病
(LSD) 中,晚期内体/溶酶体区室中胆固醇的积累导致它们的核周聚集和自噬体成熟缺陷。
自噬泡和晚期内吞体/溶酶体的运输与束缚因子的募集密切协调。具体来说,HOPS复合体可以由ARL8或 ORP1L-RAB7-RILP 复合体募集,这可以直接发生或通过 PLEKHM1。这允许顺行和逆行运输和融合事件
的
耦合。
自噬空泡的定向运输在极化神经元细胞中尤为突出,其中在远端突触末端形成的自噬小体经历长程逆行运输至胞体。自噬体的成熟是紧密结合其动力蛋白驱动的运输到细胞体,这需要骨架蛋白的协调动作,包括JNK相互作用蛋白1(JIP1),HTT相关蛋白1(HAP1)和JIP3连接的。这些因素将自噬泡与动力蛋白-动力蛋白复合物连接起来,它们的作用取决于位置和自噬体成熟度。JIP1 在轴突的远端部分起作用,HAP1在轴突中部起作用,而 JIP3 主要控制轴突近端部分成熟自噬泡的运动。
控制自噬小体成熟的机制是动态调节的,以允许自噬通量适应细胞的需要,并使自噬降解与外部输入整合。这涉及调节介导融合事件的关键成分的膜募集以及转录调节,这主要通过 MiT/TFE 家族转录因子TFEB和TFE3发生。在自噬小体生物发生过程中,各种应激和信号通路可以通过多种机制调节自噬体形成的启动,例如通过调节
Atg1 复合物
(哺乳动物中的 ULK1 复合物)和
Vps34 PtdIns3P 激酶复合物
的激酶活性、稳定性和组装。mTORC1是一种进化上保守的信号中枢,可感知营养状态和生长因子信号,在这里起着关键作用。营养状态和其他压力也整合到自噬小体成熟机制中,以增加对自噬通量的另一个控制水平。同样值得注意的是,控制自噬小体成熟的机制是异质的,至少表现出部分冗余,这可能允许灵活调节不同细胞类型的自噬通量。
SNARE蛋白质在不同的膜隔间之间动态移动。饥饿时,内体RAB蛋白RAB21被其GEF MTMR13激活,这进一步促进质膜定位的VAMP8易位至晚期内吞体/溶酶体(图
3a
)。
SNARE蛋白动力学是内溶酶体结构与自噬液泡的有效融合所必需的,如LSD所证明的,例如多种硫酸酯酶缺乏症和IIIA型粘多糖贮积症,其中SNARE被隔离在内溶酶体膜的富含胆固醇的区域并锁定在组装的复合物中。因此,融合后SNARE复合物的分解及其分类和再循环回靶膜受到损害,因此,溶酶体与内吞和自噬囊泡的融合减少。
STX17的SNARE活性受其SNARE结构域的乙酰化调节,这是一种由组蛋白乙酰转移酶 CREBBP/CBP和脱乙酰酶HDAC2控制的修饰(图
3a
)。
饥饿或mTORC1抑制使CREBBP 失活,同时促进STX17的脱乙酰化。STX17的去乙酰化促进了STX17-SNAP29-VAMP8复合物的组装,也增强了其与HOPS复合物的结合,从而促进了应激条件下的自噬体-溶酶体融合(图
3a
)。
SNAP29包含两个反平行螺旋束,是自噬小体成熟的关键SNARE 。SNAP29是翻译后修饰通过
ø -GlcNAcylation
在多个丝氨酸/苏氨酸残基,通过O-连接的β-催化的过程Ñ乙酰氨基葡萄糖(ø -GlcNAc)转移酶(OGT)。SNAP29的O -GlcNAcylation 减弱了 SNARE 复合物的组装(图
3a
)。
OGT敲低或O -GlcNAcylation 缺陷 SNAP29的表达促进含有 SNAP29的SNARE复合物的形成并促进自噬通量。UDP-GlcNAc(O -GlcNAc 添加的供体)的水平对葡萄糖、脂肪酸、尿苷和谷氨酰胺的可用性有反应。SNAP29 O- GlcNAcylation 水平会因哺乳动物细胞和蠕虫的饥饿而降低,从而将营养状态与自噬体成熟结合起来。
在自噬小体生物发生过程中,VPS34-beclin 1-ATG14复合物(PI3K复合物I)在ER和隔离膜上产生PtdIns3P,以募集效应物,如ATG18以启动自噬小体。自噬液泡上稳定水平的PtdIns3P也通过募集束缚因子HOPS复合物促进自噬小体成熟(图
3b
)。
VPS34-beclin 1-UVRAG 复合物(PI3K复合物II),以在内体上产生PtdIns3P而闻名,似乎也介导了自噬液泡上PtdIns3P产生。Pacer(与 UVRAG 相关的蛋白质作为自噬增强剂)通过结合 STX17和磷酸肌醇被募集到自噬泡,参与靶向VPS34-beclin 1-UVRAG 复合物。起搏器和UVRAG还招募HOPS复杂(图3b)。
Rubicon是一种RAB7效应子,与beclin 1相互作用并与VPS34复合物结合,拮抗UVRAG功能并负向调节自噬小体成熟。UVRAG-Rubicon相互作用通过mTORC1介导的UVRAG75磷酸化增强,而活性GTP结合的RAB7和Pacer与Rubicon竞争释放UVRAG,从而招募HOPS复合物并在饥饿下刺激VPS34活性(图3b)。然而,UVRAG在自噬体成熟中的作用存在争议,因为它对于果蝇脂肪细胞和某些哺乳动物细胞中的自噬小体-溶酶体融合是可有可无的。
除了PtdIns3P,PtdIns4P还参与自噬途径的多个步骤。在自噬小体生物发生过程中,PtdIns4P有助于将ULK1复合亚基ATG13募集到自噬小体起始位点。在成熟过程中,GABARAPs将磷脂酰肌醇4-激酶IIα (PI4KIIα) 募集到自噬小体以产生PtdIns4P,从而促进自噬小体-溶酶体融合。PtdIns4在内吞体/溶酶体上的积累也促进了融合事件。
PtdIns4P 的时空分布受PI4K和PtdIns4P磷酸酶SAC1的调节。SAC1是一种完整的膜蛋白,通过 COPI囊泡和
COPII囊泡
介导的运输在ER和高尔基体之间循环。ER定位的跨膜蛋白SUSR2(也称为TMEM39A)通过同时与SAC1和COPII外壳蛋白SEC23和SEC24相互作用来充当适配器,以促进SAC1的ER到高尔基体的转运。SAC1在TMEM39A敲低细胞中保留在ER上会增加晚期内吞体/溶酶体PtdIns4P水平,这可能是由于反式高尔基网络衍生的顺行囊泡中PtdIns4P水平升高和/或晚期内吞体/溶酶体PI4KIIα水平升高所致。因此,与 PtdIns4P 强结合的HOPS复合物的募集得到极大促进,随后自噬小体成熟得到增(图
3b)
。
RAB7的活性和动力学——包括它在自噬囊泡上的活性GTP结合状态和非活性GDP结合形式之间的转换——对于自噬通量的进展至关重要。RAB7活性受到称为“Armus”(也称为 TBC1D2A)的
GTP酶激活蛋白
(GAP) 的负调控,其靶向作用是通过与自噬液泡上的LC3和VPS34在内吞体上产生的PtdIns3P结合来介导的。Armus微调RAB7的核苷酸循环以促进自溶酶体形成和酸化(图 3b)。
Armus还被证明通过饥饿诱导的小GTPase RAC1失活受细胞营养状况的调节,RAC1与Armus竞争结合自噬液泡上的LC3。限制Armus募集到靶膜导致活性RAB7在自噬泡和内体上持续存在,这可能对自噬小体成熟产生多种抑制作用:
1. 降低了内吞体成熟为晚期内吞体/溶酶体所需的移动RAB7的可用性
2. 保持RAB7 效应子的高活性,例如Rubicon,其异常活性可能会阻碍自噬小体的成熟,如前所述
3. 还阻碍了多泡体中腔内囊泡的形成--一个对内吞途径很重要的过程
因此形成 amphisomes以及内吞体成熟为晚期内吞体/溶酶体。除了Armus的调节外,PI4KIIα产生的内吞体PtdIns4P转化为磷脂酰肌醇4,5-二磷酸也会使RAB7失活并导致其与晚期内吞体和PLEKHM1分离。这一步也有利于自噬小体的成熟。
TFEB和TFE3是转录因子与自噬键功能,因为它们控制涉及自噬小体形成、自噬小体成熟和溶酶体生物发生基因的一个网络的表达。它们的活性受磷酸化状态的广泛调节,受各种激酶和磷酸酶的控制,TFEB和TFE3的磷酸化抑制了它们的细胞质到细胞核的运输(图 3c)。
这种调节通过mTORC1与营养物质的可用性相结合。在营养丰富的条件下,氨基酸促进 mTORC1易位到溶酶体表面,在那里mTORC1被激活。这种与溶酶体表面相关的mTORC1 磷酸化TFEB和TFE3,为支架蛋白14-3-3创造结合位点以保留细胞质,从而防止这些因子的核易位和活性。除了营养物质,各种细胞外信号也调节TFEB/TFE3。例如,已经表明糖原合酶激酶3β(GSK3β)——它被蛋白激酶C (PKC) 灭活,它对几个信号转导级联反应——磷酸化TFEB以防止它转移到细胞核中。 为应对各种应激,TFEB/TFE3磷酸化受到抑制,核转运得到促进。TFEB和TFE3也被普遍存在的蛋白磷酸酶2A (PP2A) 和钙调磷酸酶(一种由饥饿触发的溶酶体钙释放或内
质网应激
激活的磷酸酶)主动去磷酸化。
蛋白质液-液相分离 (LLPS) 现在是一种既定的机制,用于在密闭的液体状隔室中浓缩蛋白质。LLPS还将转录因子、共激活因子、介质复合物和RNA聚合酶II划分为凝聚物,用于介导基因表达。最近表明,TFEB经历LLPS,并且相分离的TFEB斑点与Mediator复合物和目标mRNA共定位。核蛋白肌醇多磷酸多激酶 (IPMK) 直接与TFEB相互作用并陪伴它以抑制其LLPS(图
3c)
。
IPMK敲除在不改变TFEB磷酸化或核转运的情况下增加了TFEB转录凝聚物的形成。因此,IPMK敲除促进自噬体成熟并促进溶酶体的成熟和降解能力。
总体而言,导致成熟、降解的自溶酶体的融合事件存在高度可变性。该过程涉及多个可能是非连续的融合过程,包括具有不同内体和溶酶体结构的自噬体的融合,以及不同类型自噬泡本身之间的各种同型和异型融合事件。这涉及不同的栓系蛋白,它们对ATG8成员具有不同的结合亲和力,因此介导了用不同ATG8样蛋白修饰的不同自噬小体群体的成熟。
因此,具有不同内吞囊泡组织的不同细胞类型可能需要不同的系链来使自噬小体成熟。例如,PLEKHM1耗竭不会导致A549肺腺癌细胞和秀丽隐杆线虫中的自噬缺陷。参与自噬小体成熟的因子功能丧失也可能导致不同细胞类型在不同成熟阶段积累自噬泡。苍蝇Rab2的消耗导致肌肉细胞中的自噬小体和幼虫脂肪细胞中的amphisomes的积累。
为了获得降解潜力,自噬液泡必须有效酸化,这是通过与内吞体/溶酶体的进一步融合来实现的。晚期内吞体/溶酶体也表现出异质性,具有不同的表面RAB蛋白和不同的常驻水解酶。因此,有效的自噬降解需要将自噬液泡与多个晚期内吞体/溶酶体融合,这可能需要不同的圈套和系链。
值得注意的是,多个SNARE复合物和束缚因子的功能是部分冗余的,这在某些情况下,意味着可以通过促进替代成熟机制来挽救由一个因子消耗引起的自噬小体成熟受损。这通过表征与秀丽隐杆线虫中EPG-5活性丧失相关的自噬缺陷抑制因子来说明。
首先,通过减少SNAP29的O -GlcNAcylation实现的STX17-SNAP29-VAMP8复合物的增强组装抑制了epg-5突变蠕虫、EPG5缺陷细胞、缺乏VCP(也称为p97)的细胞中的自噬缺陷——这是对含有泛素货物的自噬小体成熟必不可少(另见下一节)——和Wdr45– Wdr45b双敲除细胞。
其次,由EPG5缺乏引起的自噬缺陷也可以通过增TMEM39A敲低细胞HOPS复合物的晚期内吞体/溶酶体募集或通过通过IPMK敲除促进TFEB活性来提高溶酶体功能和生物发生来抑制。
最后,epg-5中自噬小体成熟缺陷-通过表达RAB-7的GDP结合形式促进RAB-7动力学在非降解性自噬泡上的动态或通过消耗RBG-1-RBG-2复合物促进溶酶体生物发生和功能来拯救突变蠕虫— 一种GEF和GAP复合物,以促进突触传递和脂滴生物发生的RAB蛋白动力学而闻名。
RBG-1-RBG-2已成为RAB-7活性的新调节剂,其可能通过将RAB-7 GEF靶向溶酶体或通过促进其对溶酶体的活性来发挥作用。然而,这些机制未能抑制由参与自噬小体形成的基因敲除引起的自噬缺陷。
自噬小体的成熟与对生长条件高度敏感的内吞转运通路有着错综复杂的联系。控制自噬小体成熟机制的异质性和冗余性赋予动态内吞运输以维持细胞稳态的灵活性和稳健性。
降解自溶酶体的形成受损与各种人类疾病的发病机制有关,包括神经退行性疾病、癌症和肌肉疾病(肌病)。有缺陷的自噬小体成熟会导致有毒蛋白质聚集体的积累和破坏细胞稳态的受损细胞器。无功能的两性体、自溶酶体,甚至是与其他区室混杂融合产生的混合囊泡,例如再循环内体,可能会阻碍内吞运输和再循环,这也有助于疾病的发病机制。
自噬对神经元功能至关重要。它在维持轴突稳态方面的关键作用依赖于突触蛋白、蛋白质聚集体和受损线粒体的稳健降解介导的再循环。各种神经退行性疾病与自噬-内溶酶体系统的功能障碍有关,包括阿尔茨海默病、帕金森病、亨廷顿病、肌萎缩侧索硬化 (ALS) 和额颞叶痴呆 (FTLD)。值得注意的是,在这些疾病中,形成自噬体但随后积聚非降解自噬泡。这些神经退行性疾病涉及各种突变(图
4
)。
其中一些突变会影响与自噬小体成熟相交处的内吞运输。例如,影响动力蛋白-动力蛋白运动的p150亚基的突变会导致运动神经元疾病,而ESCRT-III复合亚基CHMP2B的突变会导致家族性ALS和FTLD 。
溶酶体功能受损也与神经退行性疾病的发病机制有关。当溶酶体酸化有缺陷时,溶酶体与自噬液泡的融合仍然会发生,但由此产生的自噬溶酶体是非降解的。
突变早老素1,家族性阿尔茨海默病的主要原因之一,会损害溶酶体酸化。此外,具有不正常的,非降解溶酶体,如戈谢病,多发性硫酸酯酶缺乏症,粘多糖贮积症类型IIIA和粘脂糖症IV型的LSDs,与神经变性相关。受损的溶酶体生物发生也是神经退行性疾病的一个特征。在这种情况下,疾病的蛋白质,如积累
α突触核蛋白
在帕金森病或与雄激素受体
的多聚谷氨酰胺扩展
在
X连锁脊髓与延髓肌肉萎缩
在细胞质死骨TFEB,损害溶酶体功能和自噬小体成熟。
溶酶体功能也可能因疾病蛋白引起的物理损伤而受损。疾病相关蛋白的淀粉样组装体,包括阿尔茨海默病中的Aβ和tau、帕金森病中的α-突触核蛋白和亨廷顿病中具有病理性多谷氨酰胺扩增的亨廷顿蛋白 (HTT),可以在细胞之间分泌和转运,并且在胞吞作用后,可以诱导受体细胞内溶酶体破裂。
直接参与自噬小体成熟的基因突变也会导致某些神经元群体的选择性损伤,导致神经退行性特征(图
4
)。
缺乏 Epg5 的小鼠表现出运动神经元的选择性丢失和 ALS 的关键特征。人类EPG5的隐性突变与多系统疾病Vici综合征有关。Vici综合征患者表现出神经发育和神经退行性特征,这些特征在Epg5基因敲除小鼠中重现。Vici综合征患者也表现出肌肉异常(骨骼肌肌病和心肌病),正如后面所讨论的,这与自噬小体成熟缺陷有关。
在神经细胞中,WDR45和WDR45B在自噬小体成熟过程中起到冗余作用,以将EPG5靶向晚期内吞体/溶酶体。WDR45中的从头突变导致
β-螺旋桨蛋白相关的神经变性
,以前称为儿童静态脑病伴成年神经变性。Wdr45基因敲除小鼠表现出广泛肿胀的轴突和学习记忆障碍,让人联想到β-螺旋桨蛋白相关的神经变性。最近出现了WDR45B在智力障碍中的潜在致病作用。Wdr45b基因敲除小鼠也表现出异常的运动行为和认知障碍,并在病理上表现出小脑萎缩和肿胀轴突中自噬体的积累。
值得注意的是,作用于自噬小体成熟的不同基因的突变会导致不同的神经病理学缺陷。这可能是因为这些基因在不同的神经元亚群中差异表达,或者在不同类型的神经细胞中是必需的,或者因为它们的突变导致不同类型的自噬物质积累,这些物质在不同细胞中可能不同。这些基因在内吞运输和再循环中的不同功能也可能导致不同的病理缺陷。
自噬对于保持肌肉质量和维持肌纤维完整性至关重要。溶酶体功能缺陷及其降解潜力与一组自噬性空泡性肌病有关,包括庞贝病、达农病和X连锁肌病伴过度自噬,这些都表现为非功能性自噬空泡的积累。庞贝病是由溶酶体酸性α-葡萄糖苷酶(GAA)缺乏引起的,该酶将糖原水解为葡萄糖。它的特点是自噬空泡的大量积累,特别是在骨骼肌中。达农病患者,这与溶酶体膜蛋白
LAMP2
缺乏有关,显示心肌病、肌病和可变智力低下。大量的自噬泡积聚在心肌细胞和患者佳能疾病和LAMP2缺陷小鼠骨骼肌细胞。具有过度自噬的X连锁肌病是一种儿童期发病的疾病,具有进行性空泡形成和骨骼肌萎缩,是由于液泡ATP酶组装因子VMA21缺乏导致溶酶体酸化受损引起的。
来自包涵体肌病、骨佩吉特病和额颞叶痴呆 (IBMPFD) 相关肌病患者的肌肉细胞积聚大的、泛素阳性的边缘空泡,这些空泡是未消化的自噬空泡。IBMPFD的致病基因是VCP。VCP主要因其在从膜或蛋白质复合物中提取泛素化蛋白质(通常错误折叠且易于聚集)以降解或回收的作用而闻名。VCP IBMPFD突变体VCP的敲低或表达导致含有泛素阳性内容的自噬液泡的积累,并且发现VCP对后期(初始酸化后)自噬小体成熟至关重要,但VCP促进进一步成熟的确切机制尚不清楚。VCP突变也与家族性ALS相关。
自噬以上下文相关的方式作为肿瘤抑制因子和启动子发挥作用。自噬通过在质量控制过程中起作用,例如去除受损线粒体和维持基因组稳定性来阻止肿瘤发生和肿瘤进展的早期阶段。相比之下,在晚期肿瘤或癌症治疗期间,自噬使肿瘤细胞能够在缺氧和代谢压力等恶劣条件下存活。
与此一致的是,由TFEB和TFE3活性增加引起的溶酶体活性升高——这可能是由染色体重排、基因扩增或上调以及核转运增强引起的——与各种肿瘤的发展或转移有关,例如肾细胞癌、前列腺癌、胰腺癌、非小细胞肺癌 (NSCLC) 和乳腺癌。
值得注意的是,TFE3 和TFEB还可以通过激活与肿瘤发生有关的信号通路,如Wnt和 TGFβ信号通路,促进癌症进展,至少部分独立于它们在自噬中的功能。然而,已证明TFE3和TFEB的表达和核输入增加可维持高自噬水平,以维持胰腺导管腺癌发病机制中的细胞内氨基酸库。增强的 TFEB活性还可以促进溶酶体胞吐作用,将蛋白水解酶(如组织蛋白酶)释放到细胞微环境中,从而促进细胞外基质重塑,从而刺激癌细胞侵袭和转移。
自噬小体成熟机制的扰动也可能有助于肿瘤的发生和发展。在EPG5改变已经提出参与乳腺癌和前列腺癌。EPG5表达在NSCLC临床样本中显着降低,并且EPG5敲低促进NSCLC细胞增殖和肿瘤发生。WDR45在子宫体子宫内膜癌患者中发生基因改变,并在宫颈癌发展中下调。然而,由于上文强调的自噬小体成熟机制的异质性和冗余性,单个成分的变化可能不足以驱动肿瘤发生和致瘤进展,并且自噬体成熟的改变很可能与其他致癌病变共存,进一步加重病理。
自噬通过捕获入侵病原体并将它们递送至溶酶体进行降解(这一过程称为异种吞噬)来对入侵病原体做出反应;这有助于抗原呈递以激活先天性和适应性免疫反应。病原体已经进化出多种策略来在起始和/或成熟步骤抑制自噬以逃避破坏。某些病原体甚至会阻止自噬体成熟并为了自身利益破坏产生的囊泡。
正链RNA病毒属于家庭小核糖核酸病毒科,如脊髓灰质炎病毒,鼻病毒,柯萨奇病毒B3和肠道病毒D68,利用双膜囊泡 (DMV) 作为膜支架进行复制和转录。在病毒感染的细胞中形成的DMV比常规的自噬小体小。它们表现出自噬体/两性体的标志,例如对LC3和晚期内吞体/溶酶体标志物LAMP1呈阳性,但它们进一步成熟为降解性自溶酶体被阻止。
β冠状病毒,包括小鼠肝炎病毒 (MHV)、中东呼吸综合征冠状病毒、严重急性呼吸综合征冠状病毒 (SARS-CoV) 和SARS-CoV-2,也会诱导DMV的形成,以锚定病毒复制和转录复合物。病毒RNA产物定位在DMV腔中,并通过双跨膜分子孔转运到细胞质中进行翻译和病毒体组装。
然而,典型的自噬机制,如LC3脂化系统,不是DMV形成或冠状病毒复制所必需的。相反,MHV感染细胞中的DMV与ER衍生的小囊泡有关,称为“
EDEMosomes
”,它将
ER相关降解
(ERAD) 的短期调节因子传递到晚期内吞体/溶酶体。然而,冠状病毒感染需要某些自噬蛋白。LC3装饰DMV,是MHV复制所必需的。与自噬结构不同,LC3与磷脂酰乙醇胺结合,非脂化LC3存在于MHV感染细胞的DMV上。
SARS-CoV-2(新冠肺炎病毒)感染需要参与PtdIns3P产生的自噬蛋白。对ER-局部跨膜蛋白的自噬EPG3(也称为VMP1)和TMEM41B是自噬体形成所必需的。EPG3和TMEM41B还可用于冠状病毒例如SARS冠状病毒-2,但在这些蛋白在病毒生命周期中起作用的步骤尚未被识别。在β冠状病毒感染的细胞中,复制的病毒在溶酶体内运输并通过胞吐途径释放。有太多的病毒颗粒或由病毒蛋白如SARS-CoV-2的ORF3a通过加载溶酶体的脱酸促进了从感染的细胞溶酶体的胞吐,从而病毒出口。自噬小体还可以扣押并介导脊髓灰质炎病毒,柯萨奇病毒B3和肠道病毒D68。
细菌通过吞噬作用侵入宿主细胞并驻留在含有细菌的液泡中。如果液泡膜受损,细菌会逃逸到细胞质中。自噬捕获细胞质或受损液泡中的细菌,并将它们输送到溶酶体进行破坏(通过异体吞噬)。细菌使用不同的
分泌系统
来传递效应物或毒素,以逃避自噬监视,甚至利用自噬液泡进行细胞内存活和生长。
细菌毒力因子可以通过抑制自噬诱导信号,损害自噬识别,或直接衰减自噬蛋白质的功能块自噬。例如,嗜肺军团菌效应蛋白RavZ通过作为半胱氨酸蛋白酶解偶联脂质偶联的ATG8蛋白来抑制宿主自噬。RavZ在羧基末端甘氨酸和倒数第二个芳香族残基之间切割ATG8蛋白,产生无法重新结合的ATG8蛋白。
SopF,沙门氏菌肠道亚种的效应物,鼠伤寒肠炎血清型,通过干扰V-ATP 酶与受损含菌液泡上的ATG16L的结合来削弱异种吞噬的起始。
通过溶解酶的沉积,含细菌的自噬液泡(自噬体或含细菌液泡和自噬体的融合囊泡)成熟为降解性自噬溶酶体也可以被抑制。
在巨噬细胞,含有的强毒株自噬泡结核分枝杆菌(H37Rv株)失败招募RAB为成熟为溶酶体。
含空泡分枝杆菌和鼠疫耶尔森氏菌表现出非降解的自体溶酶体且缺乏溶酶体酶的功能。
一些细菌(例如粘质沙雷菌、金黄色葡萄球菌、嗜吞噬细胞无形体和伯内特柯氏杆菌)甚至利用自噬液泡作为细胞内生长和增殖的复制壁龛。与自噬小体的融合促进了细菌驻留和复制的液泡的形成——通过促进单个含有细菌的液泡的融合和/或提供用于液泡扩张的膜——并且还为病原体提供营养。与此一致,这些病原体复制是通过自噬诱导促进,并且通过自噬抑制阻断。
病毒广泛使用抑制 STX17-SNAP29-VAMP8 的组装来防止降解自溶酶体的形成或积累自噬小体/两性体以进行复制或释放(图
5a
)。
柯萨奇病毒B3和肠道病毒介导D68 SNAP29的裂解病毒蛋白酶3C,分离两个SNARE基序,因而损害SNARE复合体的形成。人类副流感病毒3型的磷蛋白 (P) 与SNAP29结合以抑制其与STX17的相互作用,而丙型肝炎病毒在其中复制的细胞由于STX17的表达减少和周转增加而表现出水平降低。 病毒效应器也可以针对系留因子。PLEKHM1被柯萨奇病毒B3的蛋白酶3C蛋白水解靶向以将HOPS 复合物结合和LC3结合氨基末端与RAB7相互作用的羧基末端分离,从而消除其束缚功能(图
5a
)。
感染 SARS-CoV-2或表达病毒辅助蛋白ORF3a的细胞将HOPS复合物的成分隔离在晚期内吞体上。这可以防止功能性HOPS复合物与STX17相互作用,从而抑制 STX17-SNAP29-VAMP8 复合物
的
组装(图
5a
)。流感病毒A的M2蛋白通过干扰含beclin 1和含UVRAG的VPS34复合物
来
抑制自噬小体成熟(图
5a
)。细菌毒力因子也阻止含有细菌的自噬液泡的成熟和消除,但其机制尚不清楚,如图
5b
所示 。
自噬途径的不同步骤是治疗干预的潜在目标。抑制剂和自噬小体形成,包括VPS34和ULK1抑制剂或激活肽的活化剂
的Tat-BECLIN 1
,是自噬的有效调节剂,但尚未用于临床。相比之下,溶酶体活性
剂
可抑制溶酶体的活性并阻止其与自噬囊泡融合,已被用于多项临床试验。 通过靶向成熟机制的关键成分或通过靶向溶酶体和自噬小体生物发生的转录程序控制自噬小体活性来调节自噬体成熟也是调节自噬通量的潜在治疗选择,但如果想要在实际临床中使用这些方法,则需要进一步优化。
抑制和刺激自噬体成熟是探索的重要治疗途径。在许多神经退行性疾病中,自噬小体成熟受阻,因此恢复自噬通量将是一个重要的治疗选择。当需要抑制自噬时,例如在癌细胞中,重要的是要考虑必须中断自噬途径的步骤。当自噬在自噬小体形成或自噬小体成熟时被阻断时,可以观察到不同的结果。与此一致,已经表明阻断自噬小体的成熟有利于通过响应TNF相关凋亡诱导配体 (TRAIL) 的人癌细胞中的坏死性凋亡而导致细胞死亡,而阻断自噬小体的形成会触发TRAIL依赖性细胞凋亡在相同的细胞。
靶向控制自噬体成熟的机制(如ESCRT、SNARE和HOPS复合物)或靶向这些蛋白质的翻译后修饰是增加或减少自噬通量的理想选择。O-GlcNAc 转移酶的抑制剂和相反的酶 O-GlcNAcase的抑制剂可以通过调节SNAP29的O-GlcNAcylation分别刺激或抑制自噬小体成熟(表
1
)。
由于许多细胞蛋白是O-GlcNAc酰化的,因此阻断特定蛋白上的 O-GlcNAc位点需要开发提高靶向选择性的方法,例如使用适体或纳米抗体。另一种抑制自噬小体成熟的策略是使用小分子TCH-165来特异性激活SNAP29和STX17的蛋白酶体降解。抑制Rubicon是一种促进自噬的潜在方法;然而,药理学Rubicon抑制剂尚未开发。必须仔细评估该策略,因为Rubicon是LC3相关吞噬作用的正调节剂,该过程已知在许多炎症性疾病中具有保护作用。
TFEB/TFE3 的调节是在转录水平上控制自噬的明显策略。已经表明,TFEB过表达通过刺激脂肪吞噬作用在改善LSD和肥胖方面具有有益作用。然而,TFEB的慢性过度表达有利于胰腺肿瘤和NSCLC的发展。因此,开发小分子以急性刺激TFEB并利用自噬-溶酶体途径将有利于改善自噬具有防御作用的疾病。小分子可以通过调节其上游激酶或磷酸酶间接激活 TFEB(表1)。
例如,雷帕霉素通过抑制mTORC1活性或通过PKC-GSK3β级联从草本植物 Euphorbia peplus Linn中分离的化合物促进TFEB核转运。
氯喹 (CQ)、羟氯喹 (HCQ) 及其衍生物是唯一临床批准的作用于自噬小体成熟的药物(表 1)。它们单独使用或与其他药物联合使用,主要用于正在进行的肿瘤学试验,一般目标是通过阻断癌症治疗诱导的自噬来优化治疗。新一代二聚体CQ衍生物Ly05和DQ661的活性浓度低于CQ和HCQ。
CQ和HCQ通过抑制自溶酶体的水解能力来阻断自噬通量。它们增加自溶酶体区室的pH 值,从而阻断酸性蛋白酶和其他酶的活性。除了捕获H+的能力外,单体和二聚体CQ衍生物还通过抑制溶酶体酶棕榈酰蛋白硫酯酶1 (PPT1) 的活性来阻断溶酶体功能,该酶参与稳定V-ATPase亚基的溶酶体定位。因此,PPT1的抑制导致自噬抑制。此外,溶酶体功能和自噬小体的融合可以通过其他pH调节手段(例如通过调节V-ATPase)、抑制阳离子通道 TRPML1、抑制溶酶体酶或调节溶酶体膜动力学--融合和裂变--影响溶酶体数量和功能(表1)。
值得注意的是,溶酶体药物靶向所有酸性区室和其他途径,因此在某些情况下,溶酶体药物的有益作用可归因于除阻断自噬以外的机制。例如,这些药物通过改变内吞途径中信号分子(即NOTCH1)的运输和其他机制,独立于自噬阻断抑制肿瘤进展。还值得一提的是,由于参数不同,体外观察到的CQ和HCQ的活性在体内可能并不相同。例如,在病毒感染期间,体外使用的细胞类型可能无法反映病毒在体内的趋向性或对CQ的敏感性。此外,肿瘤中的酸性环境可以使溶酶体物质质子化并大大降低其细胞摄取。
自噬小体成熟是自噬途径中必不可少的步骤,可确保降解自溶酶体的形成。它增加了另一层复杂性,并提供了一个额外的节点来整合营养状态和压力,以调节自噬降解。在不同细胞类型和生长条件下,内溶酶体区室的独特组织和运输增加了自噬和内吞途径交叉点的复杂性。因此,反式圈套复合物和束缚因子与环境特异性因子协同作用,介导自噬小体与内吞囊泡和溶酶体的融合。需要进一步的研究来阐明不同的信号通路和压力如何协调自噬体的启动和成熟,以确保自噬流的有效进展,以及这些过程如何适应不同的细胞类型或病理生理环境。
自噬小体成熟被病原体广泛操纵以逃避破坏并进行复制和生长。使用自噬泡进行复制的病原体既可以激活自噬体起始,又可以阻止成熟以实现其最大积累。了解病毒蛋白和细菌毒力因子如何调节宿主自噬将有助于我们制定策略来干扰病原体-宿主相互作用,甚至恢复作为防御机制的自噬。随着耐多药细菌的进化,迫切需要这种策略。阐明自噬小体成熟缺陷和自噬体-溶酶体系统功能失调的潜在机制也是我们了解各种人类疾病发病机制的关键。靶向自噬体成熟——通过调节 SNARE、栓系蛋白及其调节剂以及溶酶体的生物发生和功能——为治疗这些疾病提供了一种有效的策略。
需要可靠地监测体内自噬通量的生物标志物和方法来检查自噬活动的时间变化并评估针对自噬小体成熟的干预措施。多种检测方法已被用于测量自噬通量和监测自噬体的成熟。
然而,这些分析中的许多难以在人类中实施。最近开发了几种方法来作为人类可靠的自噬生物标志物:
分离的外周血单个核细胞中的自噬通量分析用于测量人血样中的自噬活性。
正电子发射断层扫描可与缺氧示踪剂一起使用,以关联肿瘤中的缺氧和自噬,还可以通过使用与自噬底物结合的正电子发射断层扫描配体来衡量组织中特定自噬底物的水平。
生物体液中特定分子的水平也可用于确定组织中的自噬通量。例如,精氨酸酶1的血液水平反映了肝脏中的自噬活动
可是,这些方法的吞吐量低或只能应用于选定的细胞或组织。因此,为了筛选靶向自噬的药物,迫切需要可靠的、高通量的临床生物标志物,通过识别组织特异性循环自噬副产物和开发用于成像技术的通量探针来测量自噬活性。
 转化医学动态
转化医学动态